60
60
 4 минут
4 минут
Дата публикации: 11 февраля 2026
Проверено экспертом: 11 февраля 2026

Семейный аденоматозный полипоз толстой кишки (синонимы: диффузный семейный полипоз, болезнь Schmieden-Westhues) — аутосомно-доминантное заболевание, причиной которого является герминальная мутация гена APC (adenomatous polyposis coli), приводящая к развитию множества (>100) тубулярных и тубулярно-ворсинчатых аденом в толстой кишке с микроаденомами между ними и их 100%-ной прогрессии в колоректальный рак в молодом возрасте. При данном заболевании можно наблюдать образование полипов в верхних отделах желудочно-кишечного тракта, десмоидов и других внекишечных новообразований.
САП входит в группу так называемых генетически обусловленных мультиопуолевых синдромов, которые представляют собой отдельную группу заболеваний, характеризующихся развитием неоплазий желудочно-кишечного тракта с высоким риском развития онкозаболеваний, в том числе и внекишечной локализации. В эту группу наряду с САП входят синдром Пейтца-Егерса, синдром Каудена, ювенильный полипоз, наследственный синдром смешанного полипоза, синдром Банаяна-Райли-Рувалькаба, при которых частота и локализация аденоматозных или гамартомных полипов у пораженных пациентов значительно варьирует. Риск развития рака различных отделов желудочно-кишечного тракта и других органов так же различается. Эти синдромы могут быть условно разделены на аденоматозные и гамартомные. Многие клиницисты не придают большого значения наследственным факторам при оценке вероятности развития рака, и члены семьи, как правило, также не знают о наличии такой опасности. Но при этом клиническое распознавание данных синдромов необходимо не только из-за высокого риска смертности от малигнизации полипов, что составляет до 1% колоректального рака, но также из-за угрозы развития специфически ассоциированных внекишечных неоплазий, которые у пациентов в течение жизни манифестируют в разном возрасте. Кроме того, для носителей этих синдромов клиническое течение заболевания может сопровождаться опасными осложнениями, такими как кровотечение, инвагинация, обструкция кишечника. В настоящее время возможно проведение генетического тестирования, позволяющего выявить семьи-носители герминальных мутаций генов, предрасполагающих к специфически ассоциированным неоплазиям, что дает возможность назначить адекватный клинический скрининг и необходимое лечение, которые во многих случаях значительно отличаются от рекомендаций для общей популяции. [1-5,7]
Эпидемиология. Распространенность САП 1:10 000 новорожденных, преимущественно поражает женский пол, соотношение женщины/мужчины — 17:1, что значительно превышает таковое соотношение в общей популяции (3:1, соответственно). Характеризуется множеством полипов, малигнизация которых наступает в 100% случаев, и именно при этом синдроме возможна идентификация пораженных индивидов до появления рака. Выделяют 3 классических фенотипа САП.
Синдром Гарднера (Gardner syndrome) — фенотипический вариант САП. Характеризуется совместным проявлелнием десмоидных опухолей с неоплазиями желудочно-кишечного тракта. У данной группы пациентов отмечаются остеомы черепа, костно-хрящевые экзостозы, кортикальное утолщение трубчатых костей, аномальный прикус, а также кожные фибромы. Был описан в 1952 г. Следует отметить, что внекишечные проявления весьма вариабельны и могут развиваться до разрастания аденом в толстой кишке.[2,3,5,6,10]
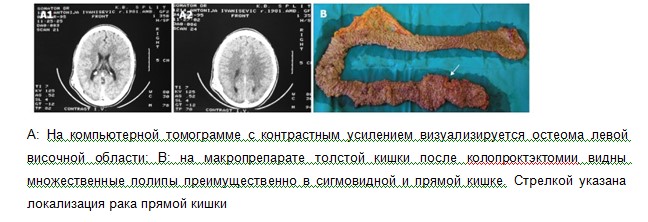
Синдром Тюрко (по имени канадского хирурга J. Turcot) так же может рассматриваться как один из фенотипических вариантов САП. Впервые описан в 1959 г., характеризуется сочетанием неоплазий толстой кишки с нейроэпителиальными опухолями головного мозга (медуллобластомой (в 80%), пинеобластомой, астроцитомой, пинеаломой или кистами шишковидной железы). Так же у пациентов с синдромом Тюрко повышен риск возникновения опухоли печени (гепатобластомы или гепатоцеллюлярного рака), которые развиваются, наряду с опухолями головного мозга, раньше полипоза толстой кишки (в 2-3 года). Следует отметить, что молекулярный патогенез синдрома Тюрко неоднороден, кроме мутации в гене APC, может быть обусловлен мутациями в генах репарации ДНК (MLH1 или PMS2), и таком случае относится к синдрому Линча, и члены выявленной семьи должны проходить молекулярный и клинический мониторинг в соответствии с рекомендациями, разработанными именно для синдрома Линча. [3,6,7]
Молекулярный патогенез САП.
Развитие САП связано с мутациями в гене АРС (Adenomatous Polyposis Coli), который идентифицирован на хромосоме 5q21 в 1991 году. Ген АРС относится к генам-супрессорам, состоит из 15 экзонов. Мутации гена АРС в большинстве случаев являются большими делециями, которые трудно выявить. Относятся к герминальным мутациям (подобные мутации присутствуют уже на стадии зиготы и передаются вертикальным путем от родителей). К настоящему времени известно более 900 мутаций. Это дает широкий спектр клинических проявлений заболевания, которые в течение жизни манифестируют в разном возрасте, а тяжесть проявления синдрома зависит от типа мутации в гене АРС. В табл. 1 приведены данные разных авторов по локализациям мутаций в гене АРС и связанные с ними возможные фенотипические проявления заболевания.
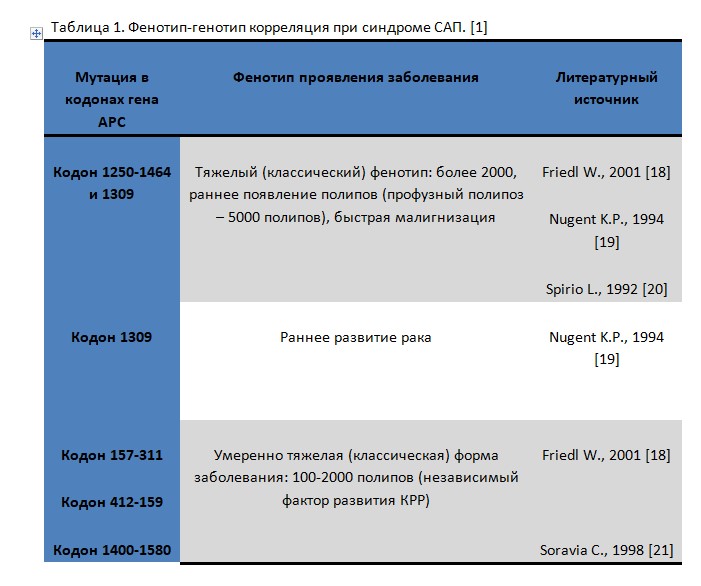
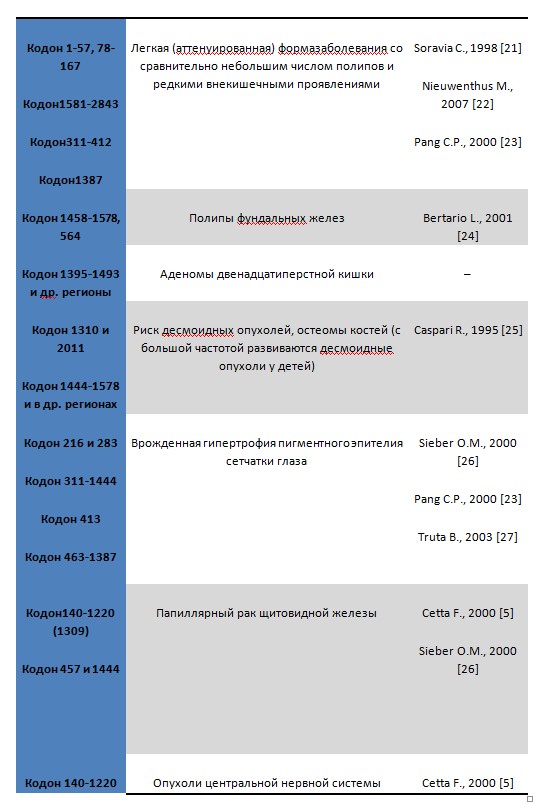

Из таблицы 1 видно, что риск развития как фенотипической формы САП, так и внекишечные проявления возможно предположить по локализации мутации в гене APC, которые на сегодняшний день изучены достаточно детально. [1-7]
В настоящее время отдельно выделяют MUTYH-ассоциированный полипоз, который наследуется аутосомно-рецессивно, и обусловленный герминальной мутацией обоих аллелей MUTYH гена. Толстокишечный фенотип имитирует АСАП, поэтому данный полипозный синдром будет рассмотрен в данном обзоре. Большинство пациентов имеют от 10 до нескольких сотен аденом. Кроме того, биаллельные герминальные MUTYH мутации были обнаружены у некоторых пациентов с ранним колоректальным раком, не ассоциированным с полипозом. В большинстве случаев у пациентов выявляются аденоматозные полипы, однако у некоторых больных данной группы неоплазии толстой кишки предсталены зубчатыми аденомами или сочетанием аденоматозных и зубчатых образований. Колоректальные полипы обычно развиваются в возрасте около 40 лет. Риск колоректального рака составляет 19% в возрасте 50 лет и 43% в возрасте 60 лет. Средний возраст малигнизации составляет 48 лет. Риск возникновения рака у родственников гетерозиготной MUTYH- мутацией сравним с таковым среди родственников первой степени родства пациентов со спорадическим колоректальным раком.
MUTYH ген состоит из 16 экзонов и расположен на хромосоме 1p34.3-р32.1. MUTYH кодирует ДНК гликозилаз, участвующих в эксцизионной репарации от 8-oxoG:есть несовпадения, вызванные окислительного повреждения ДНК.
Биаллельные MUTYH мутации встречаются примерно у 30% пациентов с 10-100 полипами и у 15% пациентов с 100-1000 полипами. У пациентов с >15 синхронными аденомами и колоректальным раком в возрасте до 50 есть очень высокий шанс выявления биаллельных MUTYH мутаций.
Молекулярный и клинический скрининг семей с САП.
Клинический диагноз САП подтверждается при колоноскопии выявлением многочисленных полипов у родственников первой линии пораженного пациента. Гететическое тестирование начинают с носителя синдрома. Других родственников тестируют в случае выявления мутации в гене APC у носителя. Риск наследования герминальной мутации составляет 50%, что характерно для любого аутосомно-доминантного заболевания. Герминальная мутация выявляется у 30-80% пациентов с САП, но у 30% она возникает de novo в результате мозаицизма. Если у пораженного полипозом пациента ни APC, ни MUTYH мутации не обнаружены, или генетическое тестирование не проводилось, родственники первой линии родства обследуются как пациенты с САП. Если же выявлены, то всем родственникам, с подозрением на САП, проводится прямое тестирование ДНК.
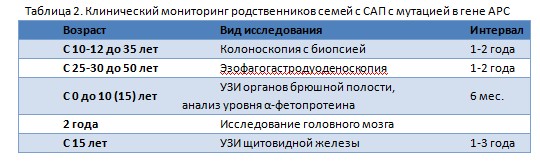
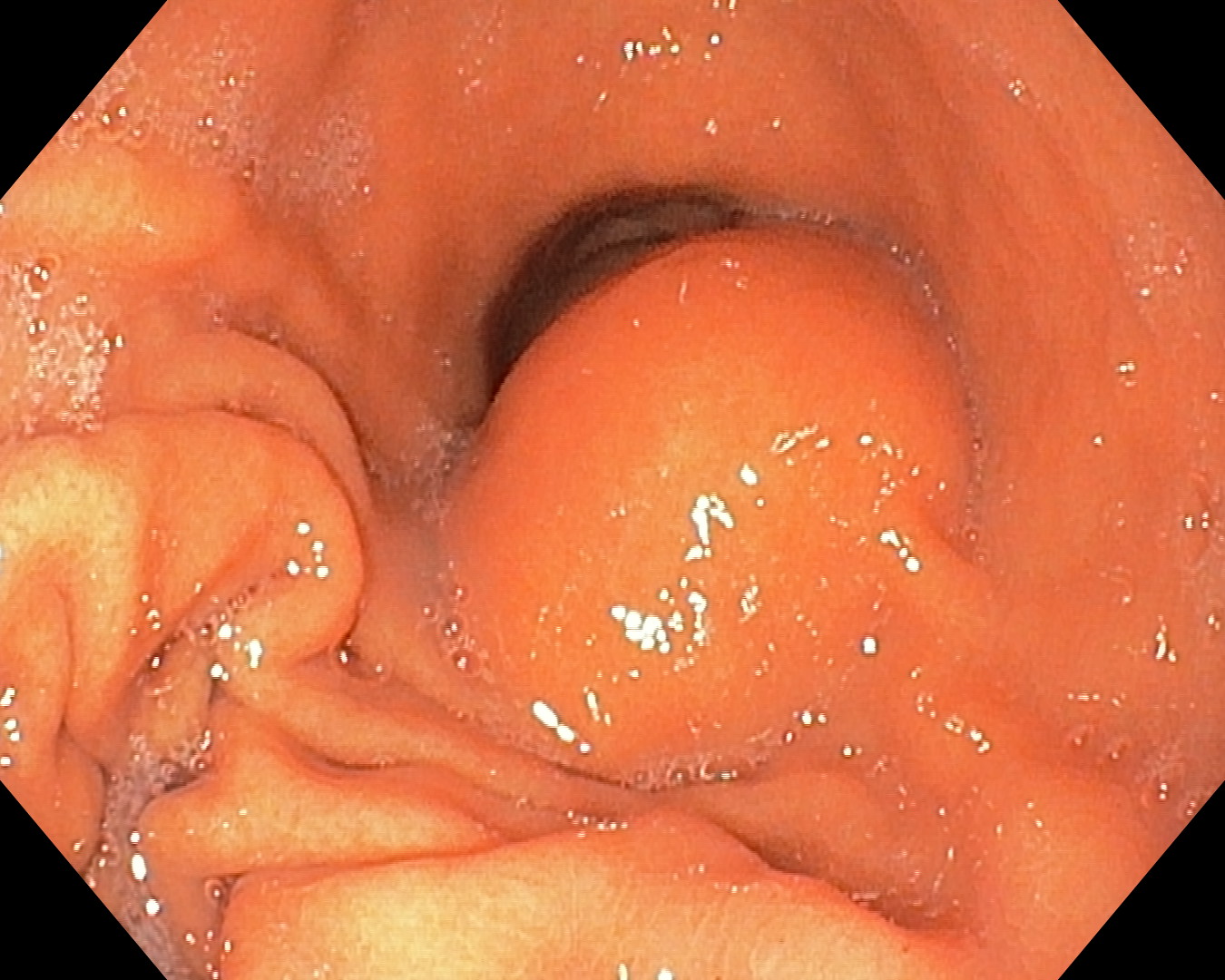
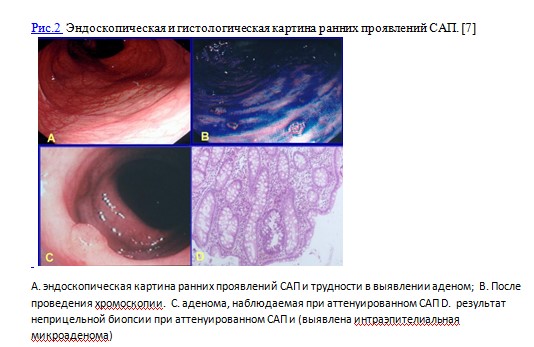
В первую очередь мониторинг САП включает ежегодную колоноскопию с биопсией начиная с 10-12 летнего возраста вплоть до 35 лет. Для родственников колоноскопию нужно начинать с пубертатного возраста или ориентироваться на такие клинические проявления как диарея, боли в животе, кровь в кале, метаболические расстройства (гипопротеинемию, гипохолестеринемию, гипокалиемию), вторичный иммунодефицит, дисбактериоз. В семьях с аттенуированной формой САП (АСАП) обследование начинают в более позднем подростковом возрасте (с 15 лет). Скрининговые колоноскопии членам семей при САП проводятся до обнаружения первых полипов толстой кишки. Во время выполнения колоноскопии необходимо описать количество полипов, их размер и распределение по отделам толстой кишки, из наиболее крупных полипов проводится биопсия. Применение вспомогательных методик визуализации (хромоскопия, узкоспектральный осмотр, осмотр с увеличением) повышает частоту выявления неоплазий толстой кишки у данной группы пациентов. (Рис.2)
Следует уделять особое внимание качеству подготовки толстой кишки к эндоскопическому исследованию. В 2014 году был проведен метаанализ, включивший 11 исследований, целью которого было сравнение малообъемного полиэтиленгликоля (ПЭГ) в комбинации с аскорбиновой кислотой (АК) —«МОВИПРЕП»®— и ПЭГ стандартного объема(4 л)в качестве препаратов для подготовки кишечника к колоноскопии. Метаанализ выявил, что частота нежелательных явлений — рвоты и тошноты — на фоне подготовки малообъемным ПЭГ в комбинации с АК по сравнению с ПЭГ стандартного объема была ниже. Согласно воронкообразному графику значимых систематических ошибок не выявлено. Таким образом, малообъемный ПЭГ в комбинации с АК не менее эффективен для очистки кишечника в качестве средства подготовки кишечника к колоноскопии, более приемлем для пациентов и характеризуется меньшей частотой нежелательных явлений по сравнению с ПЭГ стандартного объема [12]. В исследовании 2015 года, посвященном сравнению эффективности и безопасности применения комбинированного двухлитрового препарата ПЭГ с раствором электролитов и АК («МОВИПРЕП»®, Norgine) и препарата на основе пикосульфата натрия с цитратом магния («ПИКОПРЕП»®), показана более высокая эффективность первого для выявления полипов правых отделов толстой кишки и одинаковая эффективность для выявления злокачественных новообразований [11]. Таким образом, для подготовки пациентов с синдромом Линча к скрининговым колоноскопиям следует рассматривать использование малообъемного препарата полиэтиленгликоля с раствором электролитов и АК («МОВИПРЕП»®, Norgine) в сплит-дозе или одноэтапно в день исследования. Схемы подготовки в режиме приема всего объема для очистки кишечника накануне вечером использоваться не должны в связи с неудовлетворительными результатами подготовки правых отделов толстой кишки, что отражено в рекомендациях Европейского общества гастроинтестинальной эндоскопии (European Society of Gastrointestinal Endoscopy) [13]
Для хирургического лечения пациентов с САП, имеющих до 20 полипов, возможно применение эндоскопических методов удаления. В случае невозможности удаления всех полипов или развития колоректального рака больным с САП проводится оперативное лечение в объеме колэктомии или колпроктэктомии с формированием илеоректоанастомоза (илеоанального анастомоза) или постоянной илеостомы. Сроки и объем профилактического хирургического лечения продолжают обсуждаться. Считается возможным сохранение части прямой кишки после выполнения санационной резекции полипов. При этом сохраняется высокая вероятность развития метахронного рака культи прямой кишки (3,9% через 10 лет, и 25,8% через 25 лет). [1-6]
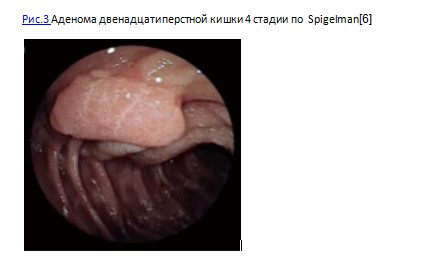
Эндоскопическое исследование верхних отделов желудочно-кишечного тракта начинают проводить с 25-30 лет и в зависимости от клинических проявлений повторяют через 2-3 года до 50-летнего возраста. У пациентов после профилактической колэктомии рак двенадцатиперстной кишки является лидирующей причиной смертности. Рак двенадцатиперстной кишки в 50% случаев развивается в парафатеральной зоне или ампуле большого дуоденального сосочка, что сопровождается панкреатитом и билиарной гипертензией. Таким образом пациентам с САП показана дуоденоскопия аппаратом с боковой оптикой. Частота развития полипов двенадцатиперстной кишки составляет 50-90%. Для оценки тяжести полипоза двенадцатиперстной кишки используется классификация Шпигельмана (Spigelman A.D. et al., 1989), согласно которой пациенту начисляются баллы за количество полипов, их размеры и гистологическое строение, потом баллы суммируются и выставляется стадия полипоза. (Таблица 3)
Таблица 3. Классификация тяжести полипоза двенадцатиперстной кишки по Шпигельману.
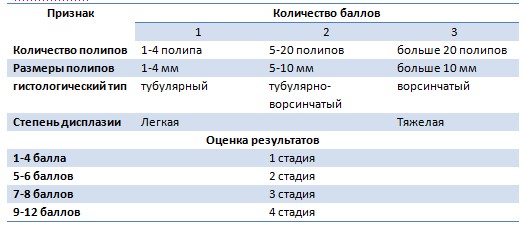
У 80% пациентов согласно данной классификации выявлялась 1-3 стадии поражения, у 20% — 4 стадия. Риск развития рака двенадцатиперстной кишки наиболее высок у пациентов с 4 стадией поражения по Шпигельману. Схожие данные были получены в скандинавско-голландских и британских исследованиях. Риск развития рака двенадцатиперстной кишки у всех пациентов с САТК составляет 4,5%. При этом риск малигнизации у больных с 3-4 стадией по классификации Шпигельмана существенно выше (7-36%) Идентификация такого рода пациентов крайне важна, поскольку позволит выявлять возможное озлокачествление на раннем этапе. При этом случаи развития рака двенадцатиперстной кишки у больных до 30 лет крайне редки. Если у пациента при эзофагогастродуоденоскопии определяется 1 стадия заболевания по классификации Шпигельмана, ему необходимо выполнение повторного эндоскопического исследования через 2-3 года, при 2 стадии – через 1-3 года, при 3 стадии – Через 6-12 месяцев, при 4 стадии необходима оценка возможности хирургического лечения.
Полипы фундальных желез, часто исчисляемые сотнями, наблюдаются у 12,5–84% пациентов с САП. Они могут покрыть всю поверхность кислотопродуцирующего эпителия и даже сливаться, придавая поверхности слизистой «матовый» вид.
Полипы фундальных желез у пациентов с САП так же могут озлокачествляться, но риск их малигнизации не превышает 0,6%. Vice-versa: при выявлении полипов фундальных желез у пациентов моложе 40 лет без длительного анамнеза приема ингибиторов протонной помпы, им следует назначить колоноскопию для исключения САП. Аденоматозные полипы желудка у пациентов с САП, проживающих в Европейских странах, развиваются относительно редко, однако среди жителей Японии и Кореи риск развития рака желудка при САП в 10 раз выше. Чаще всего образование аденоматозных полипов отмечается в антральном отделе желудка. Применение вспомогательных методик визуализации (хромоскопия, узкоспектральный осмотр, осмотр с увеличением) повышает частоту выявления неоплазий желудка у данной группы пациентов. [1-8]
Проведение капсульной энтероскопии и двухбаллонной энтероскопии для выявления полипов тонкой кишки повышает выявляемость полипов по сравнению с дуодено- и илеоскопией. [8]
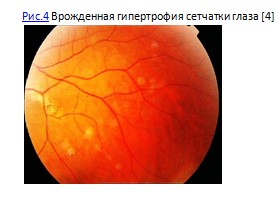
Частым компонентом САП являются доброкачественные (39-79%) и злокачественные (2-11,8%) новообразования щитовидной железы. Чаще всего поражение щитовидной железы представлено папиллярным раком, при этом часто диагностируется его крибриформная структура, которая почти не встречается в общей популяции. Следует отметить, что рак щитовидной железы манифестирует в возрасте до 28 лет, но при этом отличается более доброкачественным течением, чем спорадические случаи рака той же локализации. Исследование щитовидной железы начинают с 15 лет. Ультразвуковое исследование проводят ежегодно, при необходимости проводится тонкоигольная биопсия очаговых образований щитовидной железы. Наличие врожденной гипертрофии сетчатки глаза (Рис.4) хотя бы у одного члена семьи или мутация в гене APC с локализацией в кодоне 463-1387 указывает на повышенный риск развития опухоли щитовидной железы.
В 1,6% случаев у больных с САП развиваются гепатобластомы. Преимущественно они встречаются у мальчиков, имеющих мутации в 5´-начале гена APC в первые 5 лет жизни.
Для исключения гепатобластомы у детей проводится анализ уровня α-фетопротеина и ультразвуковое исследование органов брюшной полости в возрасте от 2 лет до 10 лет с кратностью 1 раз в 6 месяцев. У некоторых пациентов с САП риск развития гепатобластомы может сохраняться до 15 лет. Указание на гепатобластому у одного из членов семьи служит поводом для обследования пациента с 6месячного возраста с генетическим тестированием.
Обследование головного мозга следует начинать в возрасте 2 лет.
Примерно у 10-15% больных с САП могут развиваться десмоидные опухоли (десмомы). К факторам риска развития этих опухолей относятся абдоминальные хирургические вмешательства, семейный анамнез десмом, локализация мутации в гене APC после 1444 кодона. В отличие от большинства спорадических десмом, опухоли у пациентов с САП чаще всего локализуются в брюшной стенке или внутрибрюшинно. Десмомы могут быть диагностированы с помощью компьютерной томографии или МРТ. Последняя процедура также позволяет выяснить информацию об агрессивности опухоли. Кроме того, десмомы могут обнаруживаться случайно у пациентов, требующих дальнейшего хирургического вмешательства. Лечение десмоидных опухолей является комплексным и включает использование нестероидных противовоспалительных препаратов (НПВС) и/или антиэстрогенов, а также химио- и лучевую терапию, и их хирургическое удаление. При этом встречаются единичные клинические наблюдения, которые описывают спонтанную регрессию десмоидных опухолей у пациентов с САП при отсутствии какого-либо лечения. Кожные фибромы, эпидермоидные кисты, остеомы черепа, аномалии роста зубов не нуждаются в активном лечении. [2.3]
MUTYH-ассоциированный полипоз наследуется аутосомно-рецессивно. Отличается более доброкачественным течением, развитие колоректального рака отмечается в более позднем возрасте. Полипы желудка и двенадцатиперстной кишки наблюдаются у 11-17% пациентов. Риск развития рака двенадцатиперстной кишки равен 4%, имеется статистически незначимая тенденция к увеличению риска рака желудка. В качестве клинического мониторинга всем носителям мутаций в гене MUTYH показано выполнение эндоскопического обследования начиная с возраста 25 лет каждые 1-2 года, а также проведение эзофагогастродуоденоскопии с возраста 30 лет каждые 1-3 года. При этом выполнение оперативного вмешательства в виде профилактической колэктомии показано тем больным, у которых невозможно эндоскопически удалить все развившиеся аденоматозные полипы [3,5,9].
По сравнению с общей популяцией, пациенты с MUTYH-мутациями имеют почти удвоенный риск внекишечных злокачественных новообразований, включая рак яичников, мочевого пузыря, кожи и, возможно, рака молочной железы. Однако, исходя из спектра раковых заболеваний и относительно позднего возраста начала, интенсивные меры по эпиднадзору за злокачественными новообразованиями внекишечного типа не рекомендуется.[5,9]
Членам семей с САП, у которых не выявлена мутация в гене APC или MUTYH, проводится скрининг по рекомендациям для общей популяции. [1-3,5,6].
Выводы.
Семейный аденоматозный полипоз толстой кишки — аутосомно-доминантное заболевание, причиной которого является герминальная мутация гена APC (adenomatous polyposis coli), приводящая к развитию множества (>100) тубулярных и тубулярно-ворсинчатых аденом в толстой кишке с микроаденомами между ними и их 100%-ной прогрессии в колоректальный рак в молодом возрасте. Колоректальный рак, ассоциированный с САП составляет 1% от всех случаев колоректального рака. САП входит в группу генетически обусловленных мультиопухолевых синдромов, которые представляют собой отдельную группу заболеваний, характеризующихся развитием неоплазий желудочно-кишечного тракта с высоким риском развития онкозаболеваний, в том числе и внекишечной локализации, среди которых встречаются опухоли двенадцатиперстной кишки, щитовидной железы, десмоидные опухоли, гепатобластома и гепатоцеллюлярный рак, опухоли мозга и рак желудка. Диагноз основан на выявлении множественных аденом толстой кишки. Для подготовки к колоноскопии возможно использовать малолитражный ПЭГ с раствором электролитов и АК. Пациенты должны быть направлены на медико-генетическое консультирование. Риск развития как фенотипической формы САП, так и внекишечные проявления возможно предположить по локализации мутации в гене APC. В зависимости от этого членам семей с САП могут быть предложены индивидуальные программы скрининга.
Список литературы.
Оставьте свой телефон и наш оператор свяжется с вами для записи к врачу
 Город:
Город:
 Местонахождение:
Местонахождение: